Reanny Medical Devices Management Consulting Co., Ltd.
Focused · dedicated · professional
Head Office
0755-27391220
Guangzhou Company
020-82513196

Focused · dedicated · professional
Head Office
0755-27391220
Guangzhou Company
020-82513196
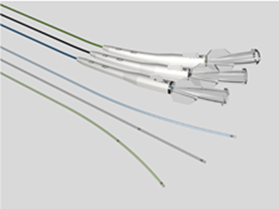
Key points for NMPA registration of microcatheters 1、 Introduction to microcathetersMicrochannels typically consist of a catheter body, radiopaque markers, connectors, and other structures. The tube body is usually thin and soft, and the surface may have a hydrophilic coating. Used for injecting diagnostic reagents (such as contrast agents), therapeutic reagents (such as drug preparations, embolic materials), and appropriate instruments (such as stents, coils) into the vascular system. Microcatheter plays an indispensable and vital role in the field of interventional treatment of coronary atherosclerotic heart disease (CHD), especially in dealing with the challenging chronic occlusive disease (CTO). As the global incidence rate of cardiovascular diseases continues to rise, and patients increasingly pursue the safety and effectiveness of minimally invasive surgery, advanced catheter technology has become a key force to promote medical progress.Microchannels, with their ultra-fine size, excellent flexibility, and precise maneuverability, perfectly meet the needs of complex coronary intervention surgeries. They can easily traverse winding, narrow, and even occluded blood vessel channels, providing doctors with unprecedented treatment pathways and achieving precise navigation and intervention of lesion sites. In the interventional treatment of complex lesions such as CTO, microcatheters not only improve the success rate of surgery, but also significantly reduce surgical risks, alleviate patient pain and recovery time.At the market level, with the continuous innovation of medical technology and the continuous expansion of clinical application scope, the demand for the global microcatheter market is showing a strong growth trend. It is predicted that this market will experience significant expansion starting from $4.3925 million in 2023, and is expected to reach a market size of nearly $8.2469 million by 2032, with a significant compound annual growth rate, reflecting the broad application prospects and huge market potential of microcatheter technology in the field of cardiovascular intervention therapy (data source from the network).In summary, as a key tool in the interventional treatment of coronary heart disease, especially in the treatment of CTO lesions, microcatheters are leading the innovation and development of cardiovascular intervention technology, bringing safer and more effective treatment options for cardiovascular disease patients worldwide.In China, the management category of microcatheters is Class III, with a classification code of 03-13-26 microcatheters. This article provides a brief introduction to the key focus points of domestic registration of microcatheters based on the "Guiding Principles for Registration and Review of Microchannels (No. 4 of 2022)" and combined with the author's previous experience.2、 Reference standards and guiding principlesYY 0285.1-2017 Intravascular catheters - Disposable sterile catheters - Part 1: General requirements;GB/T 16886 series standards;Guiding Principles for Registration and Review of Microchannels (No. 4 of 2022);2、 Requirements for Writing Overview Materials1. Product NameGenerally, "microcatheter" is used as the core word, with structural characteristics, material composition, and usage location as characteristic words, such as "coronary microcatheter".2. Structural compositionNeed to provide axial and cross-sectional views of the microcatheter; If the product is designed with a multi-layer structure, the multi-layer structure should be clearly identifiable on the cross-sectional view.3. Composition materialsFor raw materials that come into direct/indirect contact with the human body, supplier information, manufacturer information, proof of purchase documents, quality control standards, and factory inspection reports are required.For products with coatings, the coating range and method need to be provided; For hydrophilic coatings containing hyaluronic acid, the source of hyaluronic acid should be clearly identified, indicating whether it is extracted from animal tissue or prepared through microbial fermentation.3、 Non clinical data1. Product Technical RequirementsThe performance indicators in the product technical requirements should refer to Table 1 in the attachment of the Guiding Principles for Registration and Review of Microchannels (No. 4 of 2022). If the project is not applicable, the reasons should be explained.For coated products, if the coating material causes abnormal chemical performance results, it is recommended to test uncoated products to confirm whether their chemical properties are acceptable. At the same time, a comprehensive evaluation should be conducted based on the clinical application history and biocompatibility data of the coating material. If the coating can be safely used on the human body and the chemical properties of the uncoated product are normal, this chemical property may not be specified in the product technical requirements. (Tips: Chemical performance testing can be conducted only on the parts in contact with the human body. If the test results are qualified, the evaluation is also acceptable. Therefore, the safety of the product should be analyzed from multiple aspects.). )2. Product performance researchThe performance indicators listed in Appendix Table 1 of the Guiding Principles for Registration and Review of Microchannels (No. 4 of 2022) should provide determination basis and performance research data. If not applicable, reasons should be explained.The performance research data should clearly specify the sample size and the basis for determining the sample size.3. Stability studyThe shelf life verification project includes two aspects: product performance and packaging system performance, among which the product performance should include all the performance specified in the product technical requirements (especially when registering for import)4、 Clinical evaluationMicrocatheters are products listed in the "Catalogue of Medical Devices Exempted from Clinical Evaluation". If they exceed the requirements of the clinical exemption catalogue, they can refer to the "Guidelines for Technical Review of Comparative Clinical Evaluation of Intravascular Catheters of the Same Variety" for clinical evaluation or clinical trials of the same variety.
781
In China, the registration of medical devices is an important step in ensuring product safety, effectiveness, and compliance with national standards. This process is supervised and executed by the National Medical Products Administration (NMPA). The process of NMPA registration is relatively complex, covering multiple stages and detailed steps to ensure the quality and compliance of the applied products. The following is an overview of the general steps in the NMPA registration process:1、 Preparation stage1. Collect data and prepare documentsBefore applying for NMPA registration, the applicant needs to collect and prepare a series of documents and materials related to the product. These documents include but are not limited to:Basic information of the enterprise: including the company's business license, production permit, etc.Product technical documents: detailed descriptions of product technical specifications, design drawings, manufacturing processes, material composition, etc.Quality management system documents: documents that prove that the production process of the enterprise complies with the requirements of the quality management system, such as the ISO 13485 quality management system certification certificate.Clinical trial data (if applicable): High risk or new technology medical devices require clinical trial data related to product safety and efficacy.** * Product manual and label * *: Ensure that the content of the product manual and label is accurate, clear, and in compliance with Chinese regulatory requirements.2. Confirmation of Applicant QualificationsThe applicant must be a legal person or other organization established within the territory of China and have legal business qualifications. Foreign applicants need to designate a legal representative or authorized agency within China to handle the application on their behalf.2、 Application submission1. Select the registration categoryBased on the characteristics and risk level of the product, the applicant needs to determine the registration category of the medical device, namely Class I, II, or III, as well as special categories (such as in vitro diagnostic reagents, implantable medical devices, etc.). The registration requirements for different categories of products vary.2. Submit registration applicationSubmit the prepared registration application documents and related materials in accordance with the format and requirements specified by NMPA. This usually includes two methods: electronic submission and paper submission.3、 Review and Evaluation1. Technical reviewNMPA will conduct a detailed technical review of the submitted technical documents, including a review of the product's technical specifications, design, performance, and other aspects. The review process may involve experts from multiple departments and professional fields.2. Clinical evaluation (if applicable)For medical devices that require clinical evaluation, NMPA will organize or recognize third-party organizations to evaluate clinical trial data. This step aims to verify the safety, effectiveness, and clinical applicability of the product in actual use. During the evaluation process, experts will carefully analyze the design, execution process, result data, and statistical significance of clinical trials to ensure that the clinical performance of the product meets the requirements of Chinese regulations and standards.4、 On site inspection (if necessary)After completing the technical review and clinical evaluation (if applicable), NMPA may conduct on-site inspections of the production site as needed. The main purpose of on-site inspection is to verify whether the production conditions, quality management system operation, and product production process of the enterprise comply with the contents stated in the registration application documents. The inspector will focus on inspecting production equipment, raw material control, production records, quality control measures, and other aspects to ensure that the enterprise can continuously and stably produce products that meet quality standards.5、 Approval and certificationAfter comprehensive review and evaluation, if the product meets all registration requirements, NMPA will officially approve the registration application for the medical device and issue a medical device registration certificate. The registration certificate is an important document for the legal sale and use of products in the Chinese market, as well as an important symbol of the quality and compliance of enterprise products. At the same time, NMPA will publish registration information on its official website for public access and supervision.6、 Post regulationAfter obtaining the registration certificate, the enterprise still needs to accept the later supervision of NMPA. This includes regular product quality inspections, adverse event monitoring reports, and continuous supervision of the quality management system. Enterprises need to submit relevant reports and data in a timely manner in accordance with NMPA requirements to ensure that products maintain high quality and compliance throughout their entire lifecycle. At the same time, enterprises also need to pay attention to the latest regulations and standards released by NMPA, adjust and improve their quality management system and product design in a timely manner to adapt to market changes and policy requirements.
587MDSAP (Medical Device Single Audit Program) certification is a complex process aimed at simplifying and unifying the entry of medical device manufacturers into five major international markets (Australia, Brazil, Canada, Japan, and the United States) through a single audit procedure. This certification program not only improves audit efficiency, but also ensures the quality and safety of medical devices worldwide. The following is a detailed explanation of the five participating countries' audit requirements in MDSAP certification.1、 The audit requirements of the United States (FDA)The US Food and Drug Administration (FDA) has particularly strict regulations on medical devices, and its MDSAP audit requirements cover a comprehensive evaluation of the quality management system. Manufacturers need to ensure that their quality management system complies with ISO 13485 standards and additionally meets specific FDA regulations regarding medical device registration, licensing, post market surveillance, and adverse event reporting. The FDA emphasizes the importance of risk management and requires manufacturers to establish and implement effective risk management systems to identify, evaluate, and control risks in every aspect of product design, development, and after-sales service. In addition, manufacturers are required to maintain detailed records, including product testing data, production batch records, complaint handling records, etc., for FDA on-site inspections or data traceability.2、 Audit requirements for Health CanadaHealth Canada in Canada also places emphasis on the compliance of its quality management system and product safety in MDSAP audits. Manufacturers are required to submit a complete quality management system manual and documents, demonstrating how they comply with the ISO 13485 standard and detailing the process of product registration, license application, and post market supervision. Health Canada also requires manufacturers to strictly manage product design changes, production process changes, and supplier changes to ensure that these changes do not have a negative impact on product quality and safety. In addition, manufacturers need to establish and maintain an effective complaint handling system to promptly respond to and handle consumer feedback and complaints.3、 Review requirements of Japan (PMDA)The Japanese Medicines and Medical Devices Agency (PMDA) focuses on the completeness and quality of product technical documents during MDSAP audits. Manufacturers are required to submit detailed technical documents, including product design drawings, manufacturing process descriptions, material specifications, etc., to demonstrate that the product complies with Japanese technical standards and regulatory requirements. PMDA also requires manufacturers to conduct rigorous clinical evaluations and performance testing before product launch to ensure that the safety and effectiveness of the product are fully validated. In addition, PMDA has strict regulations on the identification, packaging, and labeling of medical devices, requiring manufacturers to ensure that this information is accurate and conforms to consumer usage habits and language requirements. During the PMDA audit process, manufacturers also need to pay special attention to product traceability, establish and maintain a comprehensive product traceability system, so that the source and flow of products can be quickly traced when necessary. At the same time, PMDA encourages manufacturers to adopt advanced technological means, such as blockchain technology, to improve the efficiency and reliability of product traceability.4、 Audit requirements for ANVISA in BrazilThe Brazilian National Health Surveillance Agency (ANVISA) places special emphasis on the registration and post market supervision of medical devices in MDSAP certification. Manufacturers are required to complete a detailed registration application in accordance with ANVISA's requirements, including product technical information, clinical trial data, quality management system documents, etc. ANVISA also requires manufacturers to continuously monitor the market, collect and analyze adverse event reports, take corrective and preventive measures in a timely manner after the product is launched, and ensure the quality and safety of the product. In addition, ANVISA has strict regulatory measures for the import and distribution of medical devices, requiring manufacturers to establish close cooperative relationships with authorized distributors to jointly maintain the market image and user satisfaction of their products.5、 Review requirements of Australia (TGA)The Therapeutic Goods Administration (TGA) in Australia focuses on regulatory compliance and risk management of medical devices in MDSAP audits. Manufacturers need to ensure that their quality management system complies with ISO 13485 standards and meets specific regulatory requirements of TGA. TGA requires manufacturers to conduct sufficient conformity assessment before product launch, including review of technical documents, inspection of production sites, etc. In addition, TGA emphasizes that manufacturers need to establish effective risk management mechanisms to continuously monitor and manage risks throughout the entire lifecycle of their products. For high-risk medical devices, TGA may also require manufacturers to conduct additional clinical evaluations or post market monitoring.In summary, MDSAP certification provides a unified and efficient auditing platform for medical device manufacturers, helping them better adapt to regulatory requirements in the global market and improve product quality and safety. However, due to differences in regulations, technology, and market environments among countries, manufacturers still need to have a deep understanding and meet the specific requirements of different countries when applying for MDSAP certification to ensure the success of certification and smooth market access.
563In the pharmaceutical industry, Good Manufacturing Practices (GMP) is not only the cornerstone of drug production quality management, but also an important threshold for drugs to enter the international market. The importance of GMP registration consulting services as a bridge between pharmaceutical production enterprises and regulatory agencies is self-evident. This article will delve into the key content covered by GMP registration consulting services, helping companies better understand and efficiently promote this process.1、 GMP Regulatory ConsultationThe primary task of GMP registration consulting services is to provide comprehensive and accurate GMP regulatory consultation. This includes but is not limited to the interpretation of international GMP standards (such as EU GMP, FDA cGMP) and specific regulatory requirements in each country. Consulting experts will analyze in detail the gap between the current production management system of the enterprise and GMP standards, identify potential risk points, and propose targeted improvement suggestions. In addition, with the continuous updating of regulations, consulting services also need to keep track of the latest regulatory developments to ensure that enterprises can adjust their strategies in a timely manner and meet the latest compliance requirements.2、 On site evaluation and rectification guidanceAn important part of GMP registration consulting services is conducting on-site assessments. A professional team will go deep into the production line of the enterprise to conduct a comprehensive review from multiple dimensions such as personnel training, factory facilities, equipment verification, material management, production process control, and quality assurance system. Through meticulous on-site inspections, defects and deficiencies were identified, and specific rectification suggestions were proposed based on GMP standards. During the rectification process, the consulting team will also provide continuous guidance and support to ensure the effective implementation of rectification measures until the requirements of GMP standards are met.3、 Establishment and optimization of document systemGMP registration cannot be separated from a complete document system support. Consulting services will assist enterprises in establishing or optimizing GMP related document systems, including but not limited to quality manuals, procedural documents, work instructions, record forms, etc. These documents need to provide a detailed explanation of the company's quality management policies, processes, standards, and operating procedures to ensure that every step of production activities is evidence-based and systematic. At the same time, the consulting team will also guide companies on how to effectively manage and maintain these documents, ensuring that they are timely updated, easy to access, and comply with regulatory requirements.4、 Personnel training and qualification certificationGMP requires companies to have a high-quality workforce that is capable of performing their respective job duties. Therefore, GMP registration consulting services also include personnel training and qualification certification. The consulting team will design and implement customized training plans based on the needs of the enterprise, covering multiple aspects such as GMP basic knowledge, job operation skills, quality management concepts, etc. Through training, enhance employees' professional competence and compliance awareness. In addition, for key personnel such as quality managers, production managers, etc., it is necessary to assist them in completing necessary qualification certifications, such as participating in international or domestic GMP certification training and obtaining corresponding qualification certificates. This not only enhances the personal abilities of employees, but also provides strong guarantees for the overall GMP compliance of the enterprise.5、 Simulation review and preparationIn order to ensure that the enterprise can pass the GMP audit smoothly during the formal acceptance, GMP registration consulting services will also organize simulated audits. This stage simulates the real audit process, with an experienced team of audit experts playing the role of auditors to conduct a comprehensive simulated audit of the enterprise. By simulating audits, companies can identify potential issues in advance, adjust and improve their preparation work in a timely manner, and avoid major errors in formal audits. At the same time, simulating audits is also a valuable opportunity for practice, which can help enterprise teams familiarize themselves with the audit process, enhance their confidence and adaptability in dealing with audits.6、 Follow up tracking and supportGMP registration is not a one-time process, but requires continuous attention and effort from enterprises to maintain. Therefore, GMP registration consulting services should also provide follow-up tracking and support services. This includes regular follow-up visits to enterprises to assess the operational status of their GMP system, timely identification and resolution of potential issues; Assist enterprises in responding to market changes and policy adjustments, ensuring the sustained effectiveness of their GMP compliance; And provide necessary upgrade and consulting services to help enterprises continuously improve their GMP management level and achieve sustainable development.In summary, GMP registration consulting services play a crucial role in the pharmaceutical industry. By providing professional regulatory consulting, on-site assessment and rectification guidance, document system establishment and optimization, personnel training and qualification certification, simulation audit and preparation, as well as follow-up tracking and support services, GMP registration consulting services not only help enterprises smoothly cross the GMP registration threshold, but also assist them in building and improving GMP management systems, enhancing product quality and market competitiveness, and laying a solid foundation for the long-term development of enterprises.
526In the pharmaceutical and food industries, FDA (Food and Drug Administration) factory inspections are an important part of ensuring product safety, quality, and compliance. For any enterprise that is about to undergo FDA inspection, thorough and meticulous preparation is crucial. This not only concerns the reputation of the enterprise, but also directly affects the market access and future development prospects of the product. The following is a detailed guide that outlines the key materials that companies should prepare in advance before FDA factory inspections to ensure a smooth inspection process.1、 Basic information and qualification documents of the enterpriseFirstly, enterprises should ensure that all basic registration, licensing, and certification documents are complete and up-to-date. This includes but is not limited to:Enterprise registration certificate: FDA registration certificate, confirming that the enterprise has been registered with the US FDA and obtained a unique facility identification code (FEI).Production License: A production license issued by relevant domestic or international regulatory agencies, which proves that the enterprise has the qualifications for legal production.GMP (Good Manufacturing Practice) certification: For pharmaceutical manufacturing enterprises, they are required to provide a GMP certification certificate and its latest audit report to demonstrate their quality control capabilities in the production process.Quality management system documents: including quality manuals, procedural documents, work instructions, etc., comprehensively describe the quality management system of the enterprise.2、 Product related documentsProduct documentation is a key focus of FDA inspections, and companies should ensure that all products have complete, accurate, and traceable records:Product registration and marketing authorization: For new drugs, medical devices, and food supplements, FDA approved product registration certificates or marketing authorizations are required.Product specification sheet: Detailed description of the composition, content, specifications, packaging, labels, and other information of the product.Production process flow: A detailed production process diagram, including raw material procurement, production steps, quality control points, packaging, and storage.Stability study report: Long term stability data of the product, demonstrating its quality and effectiveness under specified storage conditions.Batch production records: complete production records of recent batches of products, including raw material inspection, production operations, intermediate control, finished product inspection, etc.3、 Quality Management System Operation RecordThe FDA places great emphasis on the quality management practices of businesses, therefore, businesses should prepare sufficient records to demonstrate the effective operation of their quality management systemsDeviation and CAPA (Corrective and Preventive Action) report: Record any deviation that occurs during the production process and the implementation of its investigation, evaluation, corrective and preventive measures.*Change control records: Record the change requests, evaluations, approvals, and implementation tracking of key elements such as processes, equipment, raw materials, packaging, etc., to ensure that all changes are strictly managed and verified.4、 Employee Training and Qualification ManagementEnterprises should demonstrate their emphasis on employee training, including training records on FDA regulations, GMP requirements, product knowledge, and job operation skills. Each key position employee (such as quality manager, production supervisor, QC/QA personnel) must also have complete qualification certificates and continuing education certificates to demonstrate their professional competence and attitude towards continuous learning.5、 Supplier ManagementEffective supplier management is an important part of ensuring product quality. Enterprises should provide documents such as supplier evaluation reports, qualified supplier lists, procurement contracts, supplier audit reports, and raw material inspection reports to demonstrate their strict control and quality management of raw material sources.6、 Customer complaints and handling of adverse eventsEnterprises should establish a comprehensive mechanism for handling customer complaints and adverse events, and prepare relevant records, including complaint records, investigation reports, handling measures, and tracking results. These records not only reflect the importance that enterprises attach to consumer feedback, but also serve as an important basis for FDA to evaluate the market feedback response capability and risk management level of enterprises.7、 On site preparation and simulation exercisesBefore FDA inspection, companies should conduct comprehensive on-site cleaning to ensure a clean and orderly production environment that meets GMP requirements. At the same time, organize simulated inspections or internal audits to simulate the FDA inspection process, identify and correct potential issues, and enhance the team's ability and efficiency in responding to inspections.In summary, when facing FDA factory inspections, companies need to make comprehensive and meticulous preparations from multiple dimensions. Through detailed material preparation, standardized system operation, professional employee training, and effective supplier and customer management, enterprises can not only pass inspections smoothly, but also take this opportunity to improve their management level and market competitiveness, safeguarding product safety, quality, and compliance.
607In China, market access and continuous compliance management of medical devices are crucial tasks that directly affect public health and safety. As the main regulatory agency for medical devices, the National Medical Products Administration (NMPA) bears the important responsibility of ensuring the quality, safety, and effectiveness of medical devices. NMPA registration supervision and continuous compliance are not only strict requirements for medical device manufacturers, but also important means to ensure the healthy development of the medical market.1、 The Importance of NMPA Registration Supervision and InspectionNMPA registration supervision and inspection is an important line of defense before medical devices enter the market. This process involves a detailed review of technical documents, quality management systems, clinical trial data (if applicable), and other aspects submitted by medical device manufacturers. Technical review is a core process aimed at evaluating whether the technical specifications, design, performance, and manufacturing process of medical devices comply with Chinese regulations and technical standards. In addition, for high-risk or specific categories of medical devices, NMPA will conduct on-site audits to verify whether production facilities, quality management systems, and product manufacturing processes meet requirements. These compliance reviews during the registration process ensure that medical devices have undergone a comprehensive evaluation before being launched, minimizing potential risks to the product to the greatest extent possible. However, registration is only the beginning, and subsequent supervision and continuous compliance management are equally important.2、 Content of Continuous Compliance ManagementOnce a medical device obtains NMPA registration approval and is put on the market for sale, the manufacturer must take responsibility for ongoing compliance. This includes but is not limited to the following aspects:1. Maintenance of Quality Management System: Manufacturers must establish and maintain a quality management system that complies with the ISO 13485 standard, covering the entire process from design, procurement, production, inspection to sales and after-sales. NMPA will regularly supervise and inspect the quality management system of manufacturers to ensure its continuous and effective operation.2. Change management: In the lifecycle of medical devices, it is inevitable to encounter design changes, material changes, production process improvements, and other situations. These changes must be submitted to NMPA in advance and undergo a technical evaluation and approval process to ensure that the changed product still meets safety and effectiveness requirements. Manufacturers need to develop a detailed change management plan, evaluate the impact of changes on products, and take corresponding quality control measures.3. Annual report and regular supervision and sampling: Manufacturers holding registration certificates are required to submit an annual report to NMPA, which includes market supervision, quality feedback, safety accident reports, etc. In addition, NMPA will conduct regular supervision and sampling of registered products to verify their continued compliance. These measures help to promptly identify and address potential issues, ensuring patient safety.4. Adverse Event Reporting and Recall System: Manufacturers must establish an adverse event reporting and recall system to promptly collect, analyze, and report adverse events related to medical devices. Once serious safety hazards are discovered in the product, the manufacturer should immediately initiate a recall procedure and actively cooperate with NMPA's investigation and handling work.3、 Challenges and coping strategies facedManufacturers face many challenges in NMPA registration supervision and continuous compliance management. On the one hand, the rapid development and upgrading of medical device technology require manufacturers to continuously invest resources in product research and development and improvement; On the other hand, the continuous changes in regulations and policies have also put forward higher requirements for manufacturers' compliance management.To address these challenges, manufacturers can adopt the following strategies:1. Strengthen internal management: Establish a sound quality management system and change management system to ensure that the entire process of product design to sales complies with regulatory requirements. At the same time, strengthen internal training and education to enhance employees' compliance awareness and skill level.2. Maintain close communication with NMPA: Keep abreast of the latest regulatory policy developments and regulatory requirements to ensure that the company's compliance management is consistent with NMPA's regulatory requirements. Actively communicate with NMPA to seek guidance and assistance when encountering problems and difficulties.3. Leveraging professional expertise: Collaborating with professional medical device consultants and legal service agencies to provide comprehensive compliance consulting and service support for enterprises. These professional institutions can help businesses better understand and respond to changes and challenges in regulatory policies.4. Strengthen risk management: Establish a sound risk management mechanism and emergency response plan system to identify, evaluate, and control potential risks. Conduct comprehensive risk assessment and safety evaluation before product launch; Strengthen market monitoring and user feedback collection after product launch to promptly identify and resolve potential issues.NMPA registration supervision and continuous compliance are important means to ensure the quality and safety of medical devices. Manufacturers must fully recognize this and take corresponding measures to strengthen their compliance management work. By establishing a sound quality management system, strengthening internal management, maintaining close communication with NMPA, and leveraging professional expertise, we can address challenges and continuously improve our compliance management level. Only in this way can we ensure the legality and safety of medical devices in the market and safeguard public health.
489Head Office
Headquarters Address:Room 1509, Jingting Building, Dongzhou Community,
Guangming Street, Guangming District, Shenzhen
Phone:+86-0755-27391220
Guangzhou Company
Address:Room 506, No.3-bis, Houwangmiao Street, Shigang Road, Haizhu District, Guangzhou
Phone:+86-020-82513196
Mobile Phone:+8618127257501(WeChat Account)


Phone
0755-27391220
020-82513196
WeChat customer service
Mini Program
reanny@reanny.com