Reanny Medical Devices Management Consulting Co., Ltd.
Focused · dedicated · professional
Head Office
0755-27391220
Guangzhou Company
020-82513196

Focused · dedicated · professional
Head Office
0755-27391220
Guangzhou Company
020-82513196
In today's globalized business stage, the products and services of enterprises not only need to meet the needs of the domestic market, but also need to cross national borders and win recognition from the international market. As a large economy, the European Union has strict requirements for product quality, safety, and environmental standards. This leads to an important certification - CE certification. As a professional CE certification consultant, I am well aware of what this certification means for businesses and how to help them smoothly obtain this "pass".CE certification, also known as Conformit é Europ é enne, is a self declaration by manufacturers that their products comply with EU health, safety, and environmental protection requirements. It is not just a symbol, but also a stepping stone for enterprises to enter the European market. Products that have passed CE certification mean that they have undergone rigorous testing and evaluation and can freely circulate in the European Economic Area (EEA).For many Chinese companies, CE certification is no longer an option, but a mandatory question. With the booming development of China Europe trade, more and more Chinese products are flocking to the European market. But to stand out in this fiercely competitive environment, relying solely on price advantage is far from enough. Obtaining CE certification not only proves the quality and safety of the product, but also enhances the brand image and strengthens consumer trust.How can companies obtain CE certification? Firstly, it is necessary to clarify whether the product falls within the scope of CE certification. Different product categories correspond to different directives and standards. For example, electronic products need to comply with the Electromagnetic Compatibility Directive, while medical devices need to follow the stricter Medical Device Directive. Understanding these differences is the first step in developing certification strategies.Next, the enterprise needs to conduct product testing. This step is crucial as it directly relates to whether the product can pass certification. Testing is typically conducted by independent third-party laboratories to ensure the fairness and accuracy of the results. According to the test report, manufacturers can determine whether the product complies with relevant EU directives and coordinated standards.In addition to testing, document preparation is also an essential step that cannot be ignored. Enterprises need to prepare detailed technical documents, including user manuals, design drawings, risk assessment reports, etc. These documents will be submitted to the certification body for review to demonstrate that the company has taken necessary measures to ensure product compliance.Once the product testing is passed and the documentation is complete, the manufacturer can sign the EU Declaration of Conformity or obtain a certification certificate. This marks that the product has officially obtained the right to CE marking. However, it is worth noting that the right to use the CE mark belongs to the manufacturer, not the certification body. This means that while enterprises enjoy the convenience brought by certification, they also bear corresponding responsibilities and obligations.After obtaining CE certification, companies can leverage this advantage to vigorously promote the visibility and influence of their products in the international market. The CE mark is not only a symbol of quality assurance, but also a reflection of a company's strength and reputation. It can help businesses open the door to the European market while also enhancing their competitiveness in other international markets.CE certification can also serve as a driving force for improving the internal quality control and management level of enterprises. In order to meet the standard requirements of the European Union, enterprises must continuously improve product design, production processes, and service processes. This continuous improvement process will enable enterprises to achieve a qualitative leap in quality management, technological innovation, and brand building.CE certification is not only a necessary condition for enterprises to enter the European market, but also an important tool to improve product quality and enhance brand reputation. As a professional CE certification consultant, I am committed to helping businesses understand and meet the standards and requirements of the European Union, and smoothly complete the certification process. Let's work together and embark on the internationalization journey of our enterprise!
491In today's society, health and medical safety have become the focus of public attention. In this context, FDA (Food and Drug Administration) inspections of pharmaceutical factories are particularly important. These inspections are not only a supervision of the drug production process, but also an important means to ensure the safety and effectiveness of drugs.The core purpose of FDA factory inspections is to verify whether drug production complies with established quality standards and regulatory requirements. This process includes a review of various aspects such as production equipment, raw materials, production processes, quality control, and employee training. Through these strict inspections, the FDA can ensure that the drugs circulating on the market are safe and effective, thereby protecting consumers' health from harm.With the advancement of technology and people's increasing awareness of health, the FDA's inspection standards are also constantly improving. This means that pharmaceutical companies must invest more resources and energy to meet increasingly stringent regulatory requirements. For example, in order to ensure data transparency and traceability, many pharmaceutical companies have begun to adopt advanced information technology systems to manage their production and supply chain processes.The high standards of regulation have also brought significant challenges to the pharmaceutical industry. On the one hand, enterprises need to constantly update their equipment and technology to adapt to new regulatory requirements; On the other hand, they also need to train their employees to ensure that they understand and are able to execute the latest operating procedures. All of these require a significant amount of time and financial investment.To address these challenges, pharmaceutical companies have adopted a series of strategies. Firstly, they have strengthened communication and cooperation with regulatory agencies in order to better understand regulatory requirements and adjust their production practices in a timely manner. Secondly, they invest in research and development, constantly innovating to improve the safety and effectiveness of their products. In addition, the company also pays attention to internal quality management and has established a sound quality management system to ensure that each batch of drugs can meet the predetermined standards.While ensuring public health, the pharmaceutical industry is also striving to promote sustainable development. This includes adopting environmentally friendly production technologies to reduce the impact on the environment; Optimize supply chain management and improve efficiency; And carry out social responsibility projects to give back to the community. These measures not only help enhance the social image of enterprises, but also lay the foundation for the long-term development of the industry.FDA factory inspections play a crucial role in ensuring the safety and efficacy of drugs. Faced with strict regulatory standards, the pharmaceutical industry needs to constantly adapt and innovate to maintain competitiveness. At the same time, enterprises should actively assume social responsibility and promote the sustainable development of the entire industry. Through these efforts, we can look forward to a safer and healthier future.In this process, every step cannot be ignored. From the selection of raw materials to the production of finished products, every step needs to be strictly monitored. Only in this way can we ensure that every pill that ultimately reaches the patient's hands is safe and reliable. And behind all of this, it is inseparable from institutional arrangements such as FDA factory inspections, which are like invisible hands guiding the entire industry towards higher standards.In the future, with the deepening of globalization and technological advancements, the standards for FDA factory inspections may become increasingly high, but this is also a commitment to public health. Pharmaceutical companies and regulatory agencies need to work together to face challenges, continuously optimize and improve the regulatory system, and ensure that everyone can use safe and reliable drugs. This is not only the protection of individual health, but also a contribution to the overall well-being of society.
380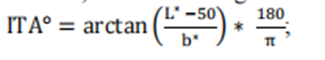
2025 version Pulse Oximeters for Medical Purposes - Non-Clinical and Clinical Performance Testing, Labeling, and Premarket Submission Recommendations Draft Guidelines and 2013 Edition Pulse Oximeters – Premarket Notification Submissions [510(k)s] Guidance for Industry and Food and Drug Administration Staff Differences and main changes in the guidelines In early 2025, the FDA released the draft guidelines for "Pulse Oximeters for Medical Purposes - Non Clinical and Clinical Performance Testing, Labeling, and Premarket Submission Recommendations" (hereinafter referred to as the 2025 draft). The clinical performance testing requirements in the draft guidelines are consistent with the guidelines for "Pulse Oximeters - Premarket Notification Submission [510 (k) s] Guidance for Industry and Food and Drug Administration Staff" (hereinafter referred to as the 2013 guidelines) released in March 2013. Compared to others, there are significant differences, and the main points of change between the two can be summarized as follows: The main differences between the 2025 draft guidelines and the 2013 draft guidelines 1. The number and diversity requirements for clinical research participants have significantly increased Old version guidelines: Only 10 subjects are required to participate in clinical studies. New draft: Require at least 150 participants and cover diverse characteristics such as skin color (evaluated using Monk Skin Tone Scale), blood oxygen saturation level (70% -100%), age, gender, BMI, and ethnicity. Each participant is required to provide at least 20 pairs of blood oxygen data, totaling at least 3000 pairs of data. Purpose: To enhance the representativeness of the sample and reduce measurement bias caused by differences in skin color, especially among people with dark skin. 2. Standardization of Skin Color Assessment Methods New draft: New requirements have been added, introducing subjective Monk Skin Tone Scale (MST) and objective Individual Type Angle (ITA) calculation methods to systematically classify and group subjects based on their skin color. The study needs to ensure that the proportion of participants in different MST groups reaches at least 30%. Old version guidelines: Failure to specify skin color assessment methods may result in insufficient coverage of clinical data on skin color. 3. Refinement of data distribution and stratification New draft: It is required that clinical data be evenly distributed in different blood oxygen saturation ranges (such as 70% -80%, 80% -90%, 90% -100%) and different skin color groups, with each range covering at least a certain proportion of the data volume. For example, each blood oxygen saturation interval needs to include at least 30% of participants. Old version guidelines: No specific requirements for data layering have been proposed, which may affect the performance validation of devices in different physiological states. 4. Label declaration and transparency requirements New draft: Added label statement, if the device passes the new standard verification, its performance in different skin color groups must be clearly marked in the label, such as "suitable for dark skin groups" or "need to be used in conjunction with clinical judgment" and other warning information.Old version guidelines: This type of declaration is not mandatory, and users may not be able to accurately understand the device's usage restrictions.5. Scope of application and adjustment of product classificationNew draft: Regarding the scope of application, the new version clearly applies to specific product codes (such as DQA, NLF, OLK, etc.), while excluding some codes covered by the old version (such as OCH, PGJ, etc.).Old version guide: Failure to provide detailed classification of applicable device types may lead to ambiguity in the implementation of the guide.Summary: The core goal of the new draft guidelines is to improve the accuracy of pulse oximeters in different skin color populations, especially to address measurement bias issues caused by skin color differences in previous studies. By expanding sample diversity, standardizing evaluation methods, refining data stratification, and enhancing label transparency, the FDA aims to drive manufacturers to optimize device performance and provide more reliable clinical decision-making basis for healthcare professionals. Pulse Oximeters for Medical Purposes - Non-Clinical and Clinical Performance Testing, Labeling, and Premarket Submission Recommendations Draft Guidelines - Translation of Clinical Performance Testing Section (1) Controlled desaturation laboratory research(2) Other considerations for pulse oximeters for children under 12 years old(3) Appendix B Considerations for Printing Skin Color CardsClinical research is crucial for evaluating the safety and effectiveness of all pulse oximeter systems within the scope of this guidance, as well as ensuring no performance differences between different skin color populations.Suggestion: We suggest conducting a controlled desaturation laboratory study as described in Annex EE of the second edition of the international standard ISO80601-2-61, December 2017 (February 2018 revised edition), to determine the accuracy of blood oxygen saturation (SpO ₂). We also suggest using this study to demonstrate that there is no performance difference in the new pulse oximeter system. In addition, for pulse oximeter systems intended for pediatric populations under 12 years old, we recommend providing convenient arterial blood samples (SaO ₂, SpO ₂) for pediatric populations under 12 years old to ensure the shape, fit, and clinical performance of the sensor site.Normally, when the proposed alternative is supported by sufficient scientific evidence, we intend to consider alternative methods beyond clinical testing to demonstrate substantial equivalence. For example, when the changes or modifications made do not affect the optical link, signal processing path, and SpO ₂ algorithm, additional clinical studies may not be necessary to demonstrate substantial equivalence.If a clinical investigation is conducted to demonstrate substantial equivalence, i.e. before obtaining a 510 (k) device license, it must comply with the Medical Device Research Exemption (IDE) regulations, namely 21CFR Part 812.. Generally speaking, we believe that pulse oximeters referred to in this guideline document should be considered low-risk devices; Therefore, this study may need to comply with the simplified requirements of 21 CFR 812.2 (b). You can refer to the guidelines of the US Food and Drug Administration (FDA) titled "Research on High Risk and Low Risk Medical Devices". In addition, applicants conducting device research aimed at demonstrating substantial equivalence in the United States are required to comply with regulations regarding institutional review boards (21 CFR Part 56) and human subject protection (21 CFR Part 50), including requirements for informed consent.When submitting clinical research data conducted outside the United States to the FDA to support the IDE or pre-market submission of these devices, the requirements of 21 CFR 812.28 may apply. Part 812.28 (a) of Title 21 of the United States Code of Federal Regulations outlines the conditions under which the FDA accepts data from clinical studies conducted outside the United States to support investigational device exemptions (IDEs) or pre-market submissions. For more information, please refer to the FDA's guidelines "Accepting Clinical Data to Support Medical Device Applications and Submissions: Frequently Asked Questions".In some cases, 'Real World Data (RWD)' can be used, such as to support indication extensions or non significant performance evaluations for devices that have received 510 (k) approval. The FDA encourages manufacturers to contact the agency when they have questions about RWD. Whether an Investigational Device Exemption (IDE) is required for collecting RWD from legally listed devices depends on the specific circumstances. For example, if an approved device is used in normal medical practice, an IDE may not be required. For more information on this topic, please refer to the FDA guidelines' Using Real World Evidence to Support Medical Device Regulatory Decisions'.(1) Controlled Desaturation Laboratory Research1. Purpose/ObjectiveThe purpose of conducting an invasive controlled desaturation laboratory study is to verify the accuracy of SpO2 in pulse oximeter systems and compare it with the reference measurement values of functional SaO2 measured by CO oximeters, while demonstrating its indistinguishable performance in different skin pigments.2. Research DesignWe suggest that you conduct the study in a population of people of color consisting of 150 or more healthy participants, following the method described in Annex EE of ISO 80601-2-61 Second Edition 2017-12 (revised version 2018-02).For research registration, we suggest the following:Evaluate the frontal pigmentation of study participants using the Monk Skin Color Scale (MST) through visual assessment - this is a subjective skin color annotation system consisting of 10 levels with high inter rating reliability (see printing recommendations in Appendix B) CIELAB color space terminology;Determine the L * and b * values of forehead pigmentation in subjects using colorimetric method, calculate the individual typological angle (ITA), defined as: According to Section 3 of the FDA draft guidelines on the collection of race and ethnic data in clinical trials and studies of FDA regulated medical products, record information related to race and ethnic diversity during the recruitment period;Assign registered research participants to three specific MST groups: 1-4, 5-7, 8-10, while ensuring the following:At least 25% of participants belong to each MST group;At least 50% of participants in MST group 8-10 have an ITA of ≤ -50 ° on their forehead; andIn each MST group, at least 40% of participants are male and at least 40% are female.We suggest that you submit the scheme used for assigning skin color (MST) and evaluating interface thermal analysis (ITA) in your pre listing submission. If additional feedback is required, we recommend communicating with the US Food and Drug Administration (FDA) through the pre established communication process outlined in the "Feedback Request and Meeting for Medical Device Submission: Q Submission Plan" guidelines, in order to discuss your proposed Skin Color (MST) allocation and Interface Thermal Analysis (ITA) evaluation plan in advance before conducting the study.In addition, we recommend measuring the Interface Thermal Analysis (ITA) value of the surface in direct contact with the sensor emitter. For fingertip sensors, in order to obtain the widest range of skin pigmentation changes applicable to the sensor placement location, we recommend evaluating the interface thermal analysis (ITA) values of the sensor site on the skin surface with pigmentation in the middle of the dorsal part of the distal phalanx (near the nail epithelium, as shown in the yellow circle in Figure 1). Figure 1: Image of a fingertip We suggest that you obtain paired observations of blood oxygen saturation (SpO2) from 3000 or more pulse oximeters and blood oxygen saturation (SaO2) from carbon monoxide oximeters. We recommend that each participant provide 20 or more data pairs that cover the range of blood oxygen saturation (SaO2) between 70% and 100%, and that each (MST) group (MST 1-4, 5-7, 8-10) and each SaO2 decile range (70% ≤ SaO2<80%, 80% ≤ SaO2<90%, 90% ≤ SaO2 ≤ 100%) account for at least 30% of the data pairs. We suggest that you provide a list of data pairs according to the participants.For more information on the principles of preclinical clinical study design that play a critical role in determining the substantial equivalence, safety, and efficacy of medical devices, please refer to the guidelines of the US Food and Drug Administration (FDA) titled "Design Considerations for Critical Clinical Studies of Medical Devices".3. Inclusion/exclusion criteriaWe recommend that your participants are healthy adults who can tolerate the desaturation described in Annex EE of ISO 80601-2-61 Second Edition 2017-12 (revised version 2018-02). In addition, we recommend excluding participants with uneven skin color on the sensor area or forehead.4. Participant demographicsWe suggest that the study population used to determine the accuracy of SpO2 should include different participants continuously selected from the existing pool of healthy participants, rather than participants from the same device calibration curve development study.We believe that the collection and presentation of race and ethnicity data should typically be submitted to the FDA prior to marketing, as described in the draft FDA guidelines on "Collection of Race and Ethnicity Data in FDA Regulated Medical Product Clinical Trials and Studies".You should describe the characteristics of the participating population that may affect the research results, including:Age;Gender;Body mass index;Race reported by self/caregiver;Race reported by self/caregiver;The MST and ITA values of each participant's forehead;ITA value of transmitter sensor position;The anatomical applicable size range of the sensor site;Study the percentage modulation range of (SaO2, SpO2) by participants when obtaining data; andThe percentage of each MST group that can tolerate complete desaturation (down to 70% of SaO2). For more information on the evaluation and reporting of age, race, ethnicity, and gender specific data in clinical trials of medical devices, please refer to the FDA guidelines "Evaluation of Gender Specific Data in Clinical Trials of Medical Devices" and "Evaluation and Reporting of Age, Race, and Ethnicity Specific Data in Clinical Trials of Medical Devices"5. AgreementWe suggest that you provide a percentage modulation range for study participants when obtaining data pairs (SaO2, SpO2), and describe the method of obtaining these values in the pre-market submission.In addition, we recommend conducting SpO2 accuracy testing under exercise conditions on all continuous (real-time monitoring and continuous data archiving) pulse oximeters and discontinuous pulse oximeters used under exercise conditions. We suggest including a description of the characteristics of each motion in the test report, including the amplitude, type, and frequency of the selected motion in the test, as well as the reasonableness of the methods used to specify the purpose of the device.6. Validity endpoints and dataWe suggest presenting Arms specifications that are less than 3% (accuracy range, may be specific indicator related concepts, need to be combined with specific context) in a statistically significant way, such as using a 95% confidence interval (95% CI). We recognize that accuracy is not only influenced by other factors, but also a function of participant characteristics, application site, and sensor geometry. Table 3 summarizes the recommended Arms (accuracy range) between the measured value (SpO2) and the reference value (SaO2) under normal conditions of blood oxygen saturation (SpO2) ranging from 70% to 100%. 7. Statistical analysis considerationsi. Common Main AnalysisFor key control desaturation studies, we recommend conducting a common primary analysis on the following performance indicators:1. Accuracy of SpOz for all study participants (Arms).2. SpOz deviation (average error) is a function of forehead Sa02 and MST.3. SpOz deviation (average error), as a function of Sa0z and ITA, is measured on the skin surface in contact with the device sensor transmitter.Ii. Recommended success criteriaFor common primary analysis, we recommend the following success criteria:Overall accuracy: Arms (accuracy range) is less than 3%. The overall accuracy must meet this requirement.Non differential performance evaluation 1: In pairwise comparisons of MST groups 1-4, 5-7, and 8-10, for the interval of 70% ≤ SaO ₂ ≤ 85%, the maximum difference in SpO ₂ deviation should be less than 3.5%; For the range of 85%<SaO ₂ ≤ 100%, the maximum difference in SpO ₂ deviation should be less than 1.5%.Non differential performance evaluation 2: When there is a 100 point change in the interface thermal analysis (ITA) at the transmitter sensor location, for the range of 70% ≤ SaO ₂ ≤ 85%, the difference in SpO ₂ deviation should be less than 3.5%; For the interval of 85%<SaO ₂ ≤ 100%, the difference in SpO ₂ deviation should be less than 1.5%.We suggest that all three success criteria should be presented in a statistically significant manner, either using a one-sided hypothesis test with a significance level of 2.5% (the p-value of the null hypothesis is less than 2.5%), or using a two-sided 95% confidence interval (the boundary of the 95% confidence interval means that the success criteria for the parameters are met).In order to visually describe device performance (i.e. consistency, bias, and uncertainty), the US Food and Drug Administration (FDA) recommends that in pre-market submissions, Bland Altman plots, modified Bland Altman plots, Q-Q plots, and inverse prediction plots should typically be provided. The FDA recommends that symbols or colors be used to identify MST groups (1-4, 5-7, and 8-10) when drawing these graphs. The FDA also recommends that the Bland Altman chart and the modified Bland Altman chart should include a 95% consistency range.Iii Sample sizeThe sample size of research participants should be taken as the maximum value of the sample size corresponding to sufficient testing power (recommended testing power of 80% or higher) to meet various success criteria and make it statistically significant. To achieve sufficient testing efficacy, the US Food and Drug Administration (FDA) recommends selecting 150 or more participants who meet the inclusion criteria described in Section 4 O (1) b as the sample size.The appropriate number of research participants depends on the accuracy of the pulse oximeter, the variability of the data, and the average number of paired repeated measurements (SpO ₂, SaO ₂) for each participant. We recommend that each participant perform an average of 20-24 synchronized paired repeated measurements, with a minimum of 17 and a maximum of 30 paired measurements per participant. Additionally, at least 30% of the data pairs in each decile range of blood oxygen saturation (SaO ₂) (70% ≤ SaO ₂<80%, 80% ≤ SaO ₂<90%, 90% ≤ SaO ₂ ≤ 100%) should be included.When there is uncertainty regarding data variability or pulse oximeter accuracy, it may be advantageous to conduct an adaptive study that can adjust the sample size based on accumulated data, if feasible.Iv. Analyze the population and methodsPerformance indicators should be analyzed using the intention to diagnose (ITD) analysis population, which is defined as all participants included in the study and all paired repeated measurement data (SpO ₂, SaO ₂), even if one or both data are invalid, cannot be evaluated, or missing. In other words, regardless of whether the data is complete or not, participants and paired repeated measurement data should not be excluded from the analyzed population. You should report the number and proportion of incomplete data pairs.v. Missing dataMeasures to reduce missing data:We suggest that you describe the measures you plan to take during the research process to minimize the occurrence of participant dropout and missing data.Reasons for missing data recording:We suggest that you clarify the reason for missing data when it occurs, for example:Participants withdraw midway;The number of paired repeated measurements of participants is insufficient (in terms of quantity or coverage of blood oxygen saturation);Participants were excluded from the analysis;The paired repeated measurement data is incomplete (invalid or missing SpO ₂ or SaO ₂).In order to provide a complete and detailed explanation of the situation of all study participants, we suggest that you collect complete information during the study period. If there is no complete information, the data may be excluded from analysis, which may introduce analytical bias and jeopardize the conclusions that can be drawn about the substantial equivalence, safety, and effectiveness of your device.h. Grouping sensors for testingIf certain sensors have similar designs or equivalent performance, it may be appropriate to group them for testing. If sensors contain the same materials and optoelectronic components, and have equivalent sensor characteristics (such as usage location), we consider them to have similar designs. If you choose to group test sensors based on similar designs, we suggest that you indicate whether all sensors within each group contain the same materials and optoelectronic components, and describe the reasons for grouping. Generally speaking, clip on sensors and adhesive sensors should not be grouped based on similar designs, as they differ in appearance, adaptability, and functional specifications. If you choose to group test sensors based on equivalent performance, we recommend that you provide valid scientific evidence and statistical analysis to demonstrate that the test results are generalizable.(2) Other precautions for pulse oximeters suitable for pediatric populations under 12 years oldIf the pulse oximeter system is intended for the pediatric population under 12 years old, consideration should be given to data that can support the accuracy of clinical performance and related pathophysiological status of the relevant pediatric subpopulation. As mentioned earlier in this guideline, due to significant differences in the shape and fit of pulse oximeter sensors, these differences may result in varying overall accuracy of the system. Therefore, clinical performance testing of pulse oximeter systems in the adult population (see Part IV O (1)) may not be sufficient to support the clinical performance of certain pediatric subgroups, such as newborns, infants, and children under 12 years old.If the device is intended for use in pediatric populations under the age of 12, the US Food and Drug Administration (FDA) recommends that manufacturers verify the performance of the device in this population by:(1) As described in Part 4 O (1) b, evaluate the performance of the pulse oximeter system using pediatric sensors in adult participants of different skin colors;(2) Evaluate the performance of pediatric participants within a specific age range (and related clinical pathological and physiological states) based on indications and sensor placement locations.Specifically, this section emphasizes that when pulse oximeters are used in children under 12 years old, their performance in children cannot be simply inferred from adult test data. Because children and adults have differences in physiological states, sensor fit, and other aspects. The first point in the validation method proposed by the FDA is to use adult participants to preliminarily evaluate the performance of pediatric sensors, as adults have high compliance and can simulate some possible situations by selecting adults with different skin colors; The second point is to directly evaluate the performance of the pulse oximeter for the target pediatric population, taking into account that children of different age groups have different clinical, pathological, and physiological states, which can affect the performance of the pulse oximeter. For example, the physiological characteristics of newborns are significantly different from those of older children, so performance evaluation should be conducted in a specific age range of pediatric populations based on specific usage instructions and sensor placement.Although pediatric (such as neonatal) clinical studies are more representative of the intended use compared to adult controlled laboratory studies, the sampled data may not cover the entire range of blood oxygen saturation (SaO2) validated in adult controlled studies, and these data were obtained under uncontrolled conditions (such as temperature, comorbidities, asynchronous data pairs). However, we suggest that you provide data and samples from a sufficient number of participants who are evenly distributed within subgroups of the population, and you need to provide a reasonable explanation of the sample size and the range of blood oxygen saturation for the data (SaO2, SpO2).In addition, we recommend including the modulation percentage range of study participants when obtaining data pairs. If your study recruits based on skin pigmentation (i.e., it is expected that the changes in skin pigmentation at the sensor placement site in your pediatric subgroup will be greater than those in the control desaturated adult study), we recommend that you include in the pre-market submission the reported race, ethnicity, MST measurement site, MST value for each participant, as well as the interfacial thermal analysis (ITA) value at the transmitter sensor site for each relevant pediatric subgroup.For additional feedback on validating the performance of pulse oximeters for patients under 12 years of age, we strongly recommend communicating with the US Food and Drug Administration (FDA) as early as possible through the pre submission process, as described in the FDA guidelines "Feedback Requests and Meetings for Medical Device Submission: Q Submission Plan", in order to discuss methods and special considerations for supporting pediatric indications for each device. Appendix B. Considerations for Printing Monk Skin Tone Color Cards Clear defined color levels in standardized color spaces such as CIELAB should be used to support the evaluation of indistinguishable performance, as described in section O (1) b of this document. One of the available options is the Skin Color Scale (MST). The US Food and Drug Administration (FDA) recommends evaluating skin color using the Skin Color (MST) method, where the color chart is based on the L * a * b * values listed in Table B1. We recommend using a calibrated printer to professionally print color cards on suitable paper. The accuracy of the color chart should be verified using a calibrated spectrophotometer. Table B1: Skin tone (MST) defined in the CIELAB color space At present, the draft is in the FDA's public consultation period, and the consultation will end on March 10, 2025. After the official draft of the guidelines is released, the new version of the guidelines will replace the 2013 version. Blood oxygen meter manufacturers need to conduct a comprehensive evaluation of their products in accordance with the new guidelines to ensure that their performance meets the requirements of the new guidelines. Rui Enni Group has been immersed in the field of medical device pre listing consulting for many years, and has multiple successful cases of pre listing and certification of oximeters. Guangzhou Zhuyitong Pharmaceutical Consulting Co., Ltd., a subsidiary of the company, undertakes clinical CRO business and has good long-term cooperative relationships with multiple research centers. We have successfully completed multiple clinical performance verifications of oximeters through joint cooperation, helping the enterprise to complete product certification and compliance listing in relevant countries. Whether it is the clinical CRO business of Zhuyitong or the pre-market consulting business of Shenzhen Ruini, the entire team has rich experience in submitting cases before the launch of blood oxygen meters. If you have any related questions, please feel free to contact us.
411In the global medical device market, compliance and safety have always been the cornerstone of a company's foundation. With the continuous development of international trade, the Medical Device Single Audit Procedure (MDSAP) certification has become a key factor for many medical device manufacturers to expand into the international market. However, in the face of complex and ever-changing international regulations and strict regulatory requirements, how to efficiently obtain MDSAP certification and ensure the smooth circulation of products in multiple national markets has become an urgent problem for many enterprises to solve.MDSAP certification is a process aimed at simplifying the entry of medical devices into the Canadian, Australian, Brazilian, and Japanese markets. It allows regulatory agencies from participating countries to accept each other's audit results. This means that if a product passes the review of one of the countries, it will be automatically accepted by other participating countries, greatly reducing duplication of work and improving market access efficiency.To successfully obtain MDSAP certification, it is necessary to have a deep understanding of the regulations in the target market. Each country has its own specific medical device regulations and standards, such as Health Canada in Canada, Therapeutic Goods Administration (TGA) in Australia, National Health Surveillance Agency (Anvisa) in Brazil, and Medicines and Medical Devices Agency (PMDA) in Japan. These institutions not only require product quality to comply with regulations, but also demand that the production process follow a strict quality management system.Next, companies need to establish or optimize their quality management system to ensure it meets the requirements of various markets. This usually involves multiple aspects of work such as document preparation, process review, and employee training. In addition, companies need to prepare detailed technical documents, including product design, manufacturing processes, quality control, and clinical evaluation reports, to demonstrate the safety and effectiveness of their products.To help businesses better cope with this challenge, professional consultants can provide comprehensive support and services. From regulatory interpretation to document preparation, from process optimization to employee training, and finally to audit coaching, a professional team can provide one-stop solutions for enterprises to ensure their smooth passing of MDSAP certification.In addition to professional knowledge and technical guidance, sharing successful cases is also an indispensable part. By learning from the experiences of companies that have successfully obtained MDSAP certification, valuable references can be provided for companies that are about to embark on this journey. For example, some companies may improve audit efficiency by strengthening internal communication, planning resource allocation in advance, or adopting advanced technological tools.The insights of experts are equally important. Industry experts typically closely monitor the latest regulatory developments and technological advancements, and their advice can help businesses anticipate potential risks and develop corresponding response strategies. For example, for upcoming new standards or policy changes, experts can provide timely information updates and impact analysis to help companies make adjustments.In the process of implementing MDSAP certification, enterprises should also pay attention to continuous improvement. Even if certified, it doesn't mean you can rest assured. Regulatory agencies may conduct regular supervisory inspections or initiate additional audits when new security issues arise. Therefore, enterprises need to maintain continuous attention and improvement of their quality management system to ensure long-term compliance.It is worth mentioning that although MDSAP certification has brought many conveniences to enterprises, it also puts forward higher requirements for the management system and product quality of enterprises. This is not only a challenge, but also an opportunity to enhance one's own strength. Through this process, enterprises can establish more rigorous management systems, improve the quality of products and services, and thus win more trust and opportunities in the international market.MDSAP certification is an important passport for medical device manufacturers to enter the international market. By gaining a deep understanding of regulations in various countries, establishing sound quality management systems, preparing sufficient technical documents, and with the help of professional consultants, enterprises can effectively address this challenge. Meanwhile, continuous learning, improvement, and innovation will be the key to maintaining competitiveness for enterprises in the constantly changing global market.If your enterprise is facing the challenge of MDSAP certification, you may consider seeking professional consulting services. Our team has rich experience and professional knowledge, dedicated to helping your enterprise successfully pass certification and explore international markets. Contact us and let's work together to safeguard your product compliance journey.
383In today's society, with the significant improvement of people's health awareness and the rapid advancement of medical technology, high-quality, safe and effective medical devices have become an urgent demand in the market. However, facing increasingly complex registration processes and strict regulatory requirements, many medical device manufacturers have encountered numerous obstacles in bringing their products to market. It is in this context that professional medical device registration agencies are particularly important. They not only help manufacturers complete complex registration procedures, but also play a key role in ensuring that products can enter the market quickly and efficiently. This article will delve into the importance of medical device registration agents, analyze their role in promoting medical innovation and safeguarding public health, and look forward to the future development trends of the industry, providing valuable insights for relevant practitioners.The core value of medical device registration agencies lies in their professionalism and service capabilities. These institutions usually have a professional team composed of industry experts, regulatory consultants, project managers, etc. They are familiar with the laws and regulations of various countries and regions, proficient in every aspect of medical device registration, from market research and product classification in the early stage, to preparing necessary technical documents and clinical trial reports, submitting application materials and communicating and coordinating with regulatory authorities, until finally obtaining the registration certificate. The entire process must be strictly operated in accordance with the norms to ensure that every step is accurate and error free. This comprehensive service greatly shortens the time to market for products, reduces the risk costs incurred by enterprises due to unfamiliarity with processes, enables manufacturers to focus on research and development and production, accelerates the launch speed of new products, and thus meets market demand faster.Medical device registration agencies also play an indispensable role in promoting medical innovation. With the advancement of technology, new medical devices are constantly emerging, but these innovative products often require a complex and lengthy registration process to enter clinical applications. Registration agencies, with their professional knowledge and rich experience, can help companies better understand regulatory requirements, design reasonable clinical trial plans, collect and organize scientifically rigorous data, and improve the likelihood of product approval. In addition, they can provide market access strategy recommendations for enterprises based on the latest research results and technological trends, help innovative medical devices benefit patients as soon as possible, and promote technological progress and improvement of medical service quality in the entire industry.Ensuring public health is another important mission of medical device registration agencies. Medical devices are directly related to the treatment effectiveness and life safety of patients, therefore, their safety and effectiveness must be strictly verified. Registration agencies ensure that every medical device entering the market meets international and domestic quality and performance standards by participating in product testing, evaluation, and audit processes, effectively preventing unqualified products from entering the market and protecting consumer rights from infringement. At the same time, they will regularly track the usage and feedback information of marketed products, assist regulatory agencies in risk management and quality control, and continuously improve the overall level of medical devices.Although medical device registration agencies play an important role in promoting industry development, they currently also face some challenges. Firstly, the increasing demand for cross-border registration in the context of globalization has brought difficulties to unified management and coordination due to differences in regulations between different countries and regions. Secondly, with the application of new technologies such as artificial intelligence and big data, the complexity and diversity of medical devices continue to increase, which puts higher demands on the professional competence of registered personnel. Furthermore, intensified market competition may lead some agencies to resort to unfair means to compete for business, damaging the reputation of the entire industry.In the face of these challenges, the future development of medical device registration agencies should focus on the following aspects: firstly, strengthening international cooperation and communication, establishing a closer global network to meet the demand for cross regional registration; Secondly, continuously improving the professional capabilities and technical level of the internal team, especially cultivating more professional talents in emerging technology fields; The third is to strengthen industry self-discipline, establish a sound integrity system, and maintain a fair competition market environment; The fourth is to use digital tools to optimize workflow, improve efficiency while reducing costs.As a bridge connecting manufacturers and regulatory agencies, medical device registration agencies play an indispensable role in accelerating the process of medical device market launch, promoting technological innovation, and safeguarding public health. With the development of the industry and changes in social demands, these institutions need to constantly adapt to new situations, improve service quality and efficiency, in order to better serve the health cause of patients and the entire society. For companies and individuals interested in entering this field, a deep understanding of its value and the challenges it faces, as well as grasping future development trends, will be the key to success.
378In the wave of globalization, if enterprises want to establish themselves in the European Economic Area, a market full of opportunities and challenges, CE certification is undoubtedly the golden key to opening the door to success. As an expert who has been deeply involved in the field of certification consulting for many years, I am well aware of the importance of CE certification for enterprises to enter the EU market for their products.Imagine when your product is placed on the shelves of European consumers, it is not only a recognition of quality, but also a demonstration of the brand's international influence. And the premise of all of this is to obtain that small CE mark sticker - it is a symbol of safety and compliance, and the cornerstone of consumer trust. But many companies often feel confused and at a loss when facing the tedious and complex CE certification process. Don't worry, this is exactly the purpose of our professional team's existence.CE certification, also known as Conformit é Europ é enne, is an abbreviation for "European Conformity Assessment" in French. It indicates that the product meets the requirements of EU directives on health, safety, and environmental protection. Whether it is machinery, electronic appliances, or medical devices, as long as they are sold in the European market, the CE mark is like a passport that cannot be obtained without it. This is not just a symbol, but also a proof that the quality and safety of the company's products meet international high standards.Let's delve into the core value of CE certification. It ensures the free circulation of products throughout the EU, eliminates technical trade barriers, and opens up broader market space for enterprises. Products with the CE mark can reduce customs inspection procedures and accelerate market access, which is crucial for companies eager to seize market opportunities. At the same time, CE certification can significantly enhance the brand image and market competitiveness of products, boost consumer confidence, and thus increase sales and market share.How to obtain this valuable CE certification? Firstly, companies need to determine the EU directives applicable to their products, such as the Low Voltage Directive, Electromagnetic Compatibility Directive, or Medical Device Directive. Subsequently, in accordance with the requirements of relevant directives, adjustments will be made to product design, production processes, and necessary testing to ensure that the product meets all safety and performance standards. This may involve multiple stages such as document preparation, risk assessment, and establishment of a quality control system. After completing these preparations, choose a qualified third-party certification agency for auditing and testing. Once the product passes all evaluations, it will obtain CE certification and the right to use the logo.It is worth mentioning that CE certification is not a one-time solution. With product updates, iterations, or regulatory changes, companies need to continuously monitor and update the certification status in a timely manner. Therefore, choosing a professional certification consulting firm as a partner is particularly important. We can not only provide the latest regulatory interpretations, but also tailor the most suitable certification scheme for you to ensure an efficient and smooth entire process.In terms of price, our service offers extremely high cost-effectiveness. Although the specific cost may vary depending on factors such as product type, complexity, and required testing scope, we promise to provide transparent pricing without any hidden costs. More importantly, we provide not only certification services, but also follow-up market access guidance, technical support, and even brand promotion suggestions, helping you comprehensively enhance your international market competitiveness.Dear entrepreneurs, if you aspire to expand your business to the European continent and make your products shine on the international stage, then please do not let CE certification become an obstacle. Contact us and let's work together to safely and efficiently promote your products to every corner of the world. Remember, every leap is a new starting point for brand growth; Every certification is an investment in future success. In this era of globalization, let us take professionalism as the boat and service as the sail, and sail together towards a more brilliant tomorrow!
379Head Office
Headquarters Address:Room 1509, Jingting Building, Dongzhou Community,
Guangming Street, Guangming District, Shenzhen
Phone:+86-0755-27391220
Guangzhou Company
Address:Room 506, No.3-bis, Houwangmiao Street, Shigang Road, Haizhu District, Guangzhou
Phone:+86-020-82513196
Mobile Phone:+8618127257501(WeChat Account)


Phone
0755-27391220
020-82513196
WeChat customer service
Mini Program
reanny@reanny.com